Tool · Desktop · Windows · Experimental
Structural Family Classification
结构家族分类
Classify SELEX-derived DNA aptamers into structural families by their loop region, not their full-length sequence. Multi-structure suboptimal folding plus weak-helix dissolution lets two aptamers with different stems but the same loop architecture land in the same family.
面向 SELEX 适配体的环路结构家族分类——按环路区域而非全长序列分组。多结构次优折叠 + 弱螺旋融合,使茎区不同但环路结构一致的适配体落入同一家族。
v0.9.4 · Built on AptaFold + Aptamer Analysis · Loop families still under wet-lab validation — treat output as a structure-aware shortlist.
/ Introduction · 介绍
What it does.这个工具能做什么。
Conventional clustering of post-SELEX sequence pools matches aptamers by full-length string similarity — which conflates "same fold, different stem" cases. This pipeline switches the comparison to the loop region after weak-helix dissolution: every sequence is folded into up to 25 suboptimal structures, the loops are extracted from each structure, and helices with ≤ 3 bp are dissolved so flanking loops merge. The merged loops are then grouped by a purpose-built, from-scratch conserved-motif classifier — not a reuse of the Aptamer Analysis Tool's k-mer / DBSCAN engine. Instead of measuring whole-loop string distance (which dilutes a short conserved core inside a long variable loop), it discovers the conserved blocks a loop carries and groups loops that share the same block combination. There is no N×N distance matrix, so the classifier has no corpus-size ceiling and runs in seconds on a full pool.
传统 SELEX 后聚类按全长序列相似度匹配适配体——会把"折叠相同、茎区不同"的情况混淆。本流水线将比较对象切换为弱螺旋融合后的环路区域:每条序列产生最多 25 个次优结构,从每个结构中提取环路,≤ 3 bp 的弱螺旋被融合使相邻环路合并。随后用一套完全自主重写的保守 motif 分类引擎对融合后的环路分组——并非复用 Aptamer Analysis Tool 的 k-mer / DBSCAN。它不再测量整条环路的字符串距离(那样会让长可变环路里一小段保守核心被稀释),而是发现每条环路携带的保守区块(block),把拥有相同区块组合的环路归为一族。整个过程没有 N×N 距离矩阵,因此不受语料规模上限限制,全库分类只需数秒。
-
Loop-centered, not sequence-centered
环路中心,非全长中心
Two aptamers with the same loop architecture but different stems land in the same family — capturing the structural feature that actually drives binding.
具有相同环路结构但茎区不同的适配体被归为同一家族——捕捉真正决定结合特性的结构特征。
-
Multi-structure suboptimal folding
多结构次优折叠
Up to 25 suboptimal folds per sequence within
4 kcal/molof the MFE — the family the aptamer biologically samples, not just the single lowest-energy guess.每条序列在 MFE 上下
4 kcal/mol内产生最多 25 个次优结构——反映适配体生物学上实际采样的结构空间,而非只取单一最低能量结构。 -
Weak-helix dissolution merge
弱螺旋融合
Helices of
≤ 3 bp(≤ 6 paired nucleotides) are treated as part of the loop — so a real big loop chopped by a spurious 2-bp stem is recovered as one unit. Threshold is swept over0 – 5 bp.≤ 3 bp(≤ 6 个配对碱基)的螺旋视为环路的一部分,使被虚假 2 bp 茎切碎的大环路重新合并为一个单元。阈值在0 – 5 bp内可扫描。 -
Self-written motif-block classifier
自研 motif 区块分类引擎(核心创新)
Not a clustering library and not the Aptamer Analysis Tool's engine — a from-scratch classifier. It mines short enriched k-mer cores, grows each into a consensus block, then names a family by the combination of blocks a loop carries (e.g.
GCGTT+GAAGG). A member must match a block's full consensus, so background sequences stay ungrouped instead of bloating an over-broad family.不是聚类库,也不是 Aptamer Analysis Tool 的引擎——而是完全从零写的分类器。先挖掘富集的短 k-mer 核心,逐步生长为共识区块(block),再以一条环路携带的区块组合命名家族(如
GCGTT+GAAGG)。成员必须匹配区块的完整共识,背景序列保持未分组,不会撑大成过宽的家族。 -
Streams to disk, multi-core fold
流式 I/O,多核折叠
15 000 sequences × ~25 folds ≈ 375 000 folds. Folding runs one sequence per CPU core; intermediate tables are streamed to disk row-by-row, so peak RAM stays flat.
15 000 条序列 × 约 25 个折叠 ≈ 375 000 个结构。每个 CPU 核负责一条序列;中间表逐行流式写盘,峰值内存恒定。
-
Instant re-classify after one fold
折叠一次,秒级重分类
Folding is the slow stage. Once cached, changing the merge threshold, family-count knob (
min-family), motif-mismatch tolerance, or motif-extension cutoff re-runs the classify + assign stages in seconds.折叠是最慢的一步。完成后,调整融合阈值、家族数量(
min-family)、motif 错配容忍度或 motif 延伸阈值,均可在秒级内重跑分类与归属。
/ Pipeline · 流水线
15 000 trimmed sequences
│
▼ Fold — reuses AptaFold's seqfold / ViennaRNA suboptimals
02_fold_predictions.csv
│
▼ Loop extraction (self-written)
03_raw_loops.csv dot-bracket → pair table → loops
04_merged_loops_bp3.csv weak-helix dissolution (≤ 3 bp)
05_loop_map_bp3.csv loop ↔ (seq, fold, span, ΔG)
06_loop_corpus_bp3.fasta de-duped loop strings + recurrence
│
▼ Motif-block classifier (self-written — the core engine)
· mine enriched k-mer cores → grow consensus blocks
· family = the block COMBINATION a loop carries
· strict full-consensus membership; no N×N matrix
│
▼ Assign + post-process (self-written)
· dual-anchor merge — fuse families sharing a 5′ AND 3′ framework
· ungrouped rescue — recover high-read near-identical winners
07_aptamer_family_bp3.csv per-aptamer family
08_family_representatives_bp3.csv picks for validation
/ Usage · 使用方式
How to use it.使用流程。
The whole pipeline is wrapped in one desktop GUI (启动GUI.bat on Windows). Folding is launched as a separate subprocess so the GUI stays responsive throughout.
整套流水线封装在一个桌面 GUI 中(Windows 上 启动GUI.bat 一键启动)。折叠任务在独立子进程中运行,主界面始终保持响应。
-
01
Launch & load sequencing data
启动并导入测序数据
Open the GUI. Pick a FASTQ file (will be trimmed) or an already-trimmed FASTA / CSV from Aptamer Analysis Tool. Read counts are preserved if the FASTA headers carry
count=N.打开 GUI,导入 FASTQ 文件(自动 trim)或 Aptamer Analysis Tool 已 trim 的 FASTA / CSV。如 FASTA 头部含
count=N,read 数会被保留。
Fig. 01 — Main window · 主窗口(导入 / Trim / 折叠 / 家族分类四组参数) -
02
Configure trim, folding & family parameters
设置 trim、折叠与家族参数
In the four parameter panels: pick a trim mode and library length
N; pick a folding engine (viennarnarecommended, salt-sensitive, ~20–30× faster than alternatives); set[Na⁺] / [Mg²⁺] / [K⁺] / [Ca²⁺]and temperature to match your selection buffer; set the loop merge thresholdbp(auto by default), target family count, and motif strictness.在四组参数面板中:选择 trim 模式与文库长度
N;选择折叠引擎(推荐viennarna,盐离子敏感,比其他引擎快约 20–30 倍);设置[Na⁺] / [Mg²⁺] / [K⁺] / [Ca²⁺]与温度以匹配筛选缓冲液;设置环路融合阈值bp(默认 auto)、目标家族数量、motif 严格度。 -
03
Run the pipeline
运行流水线
Folding (Stage 1/4) is the slow part — every sequence ×
fold_budget≈140constrained refolds, run in parallel across all CPU cores. Stages 2/4 (loop extraction + merge), 3/4 (motif-block classification), and 4/4 (assign + dual-anchor merge + rescue) each finish in seconds once folding is cached. The bottom-left console reports each stage's row counts and the run's headline stats (aptamers, total folds, loop corpus size, families, grouped fraction, representatives).折叠(阶段 1/4)是最慢的一步——每条序列 ×
fold_budget(约140次受限重折),跨所有 CPU 核并行。一旦折叠结果缓存,阶段 2/4(环路提取与融合)、3/4(motif 区块分类)、4/4(归属 + 双锚点合并 + 救回)均秒级完成。左下角控制台逐阶段汇报行数与运行统计(aptamers、total folds、loop corpus、families、grouped 比例、代表序列数)。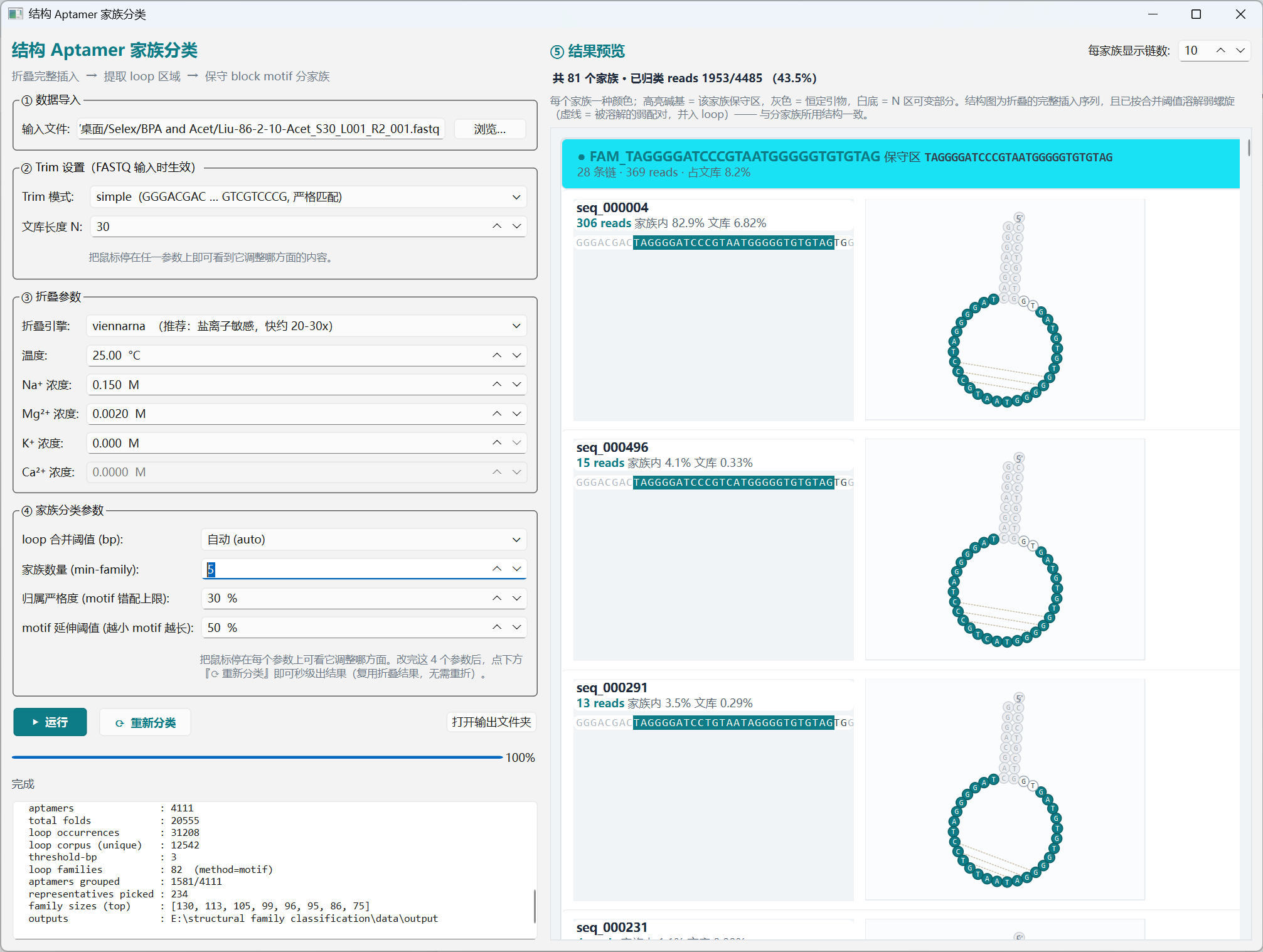
Fig. 03 — Pipeline complete · 流水线完成(4111 aptamers · 82 families · 1581 grouped · 234 reps) -
04
Review families & iterate
查看家族结果并迭代
The result panel renders each family in a distinct colour: highlighted bases = the family's conserved motif, gray = primer flanks, white = variable N region. Each chain card also draws the full-insert 2D secondary structure (dissolved at the merge threshold), with the conserved motif painted in the family colour. Tweak the merge bp, family count (
min-family), motif-mismatch tolerance, or motif-extension cutoff and click Re-classify — the fold cache is reused, so a new family palette appears in seconds.结果面板按家族用不同颜色显示:高亮碱基 = 该家族保守 motif,灰色 = 引物恒定区,白底 = N 区可变部分。每条 chain 卡片同时画出完整插入序列的 2D 二级结构(按融合阈值溶解),保守 motif 用家族颜色高亮。调整融合 bp、家族数量(
min-family)、motif 错配容忍度或 motif 延伸阈值后点击 重新分类,复用折叠缓存,秒级出新结果。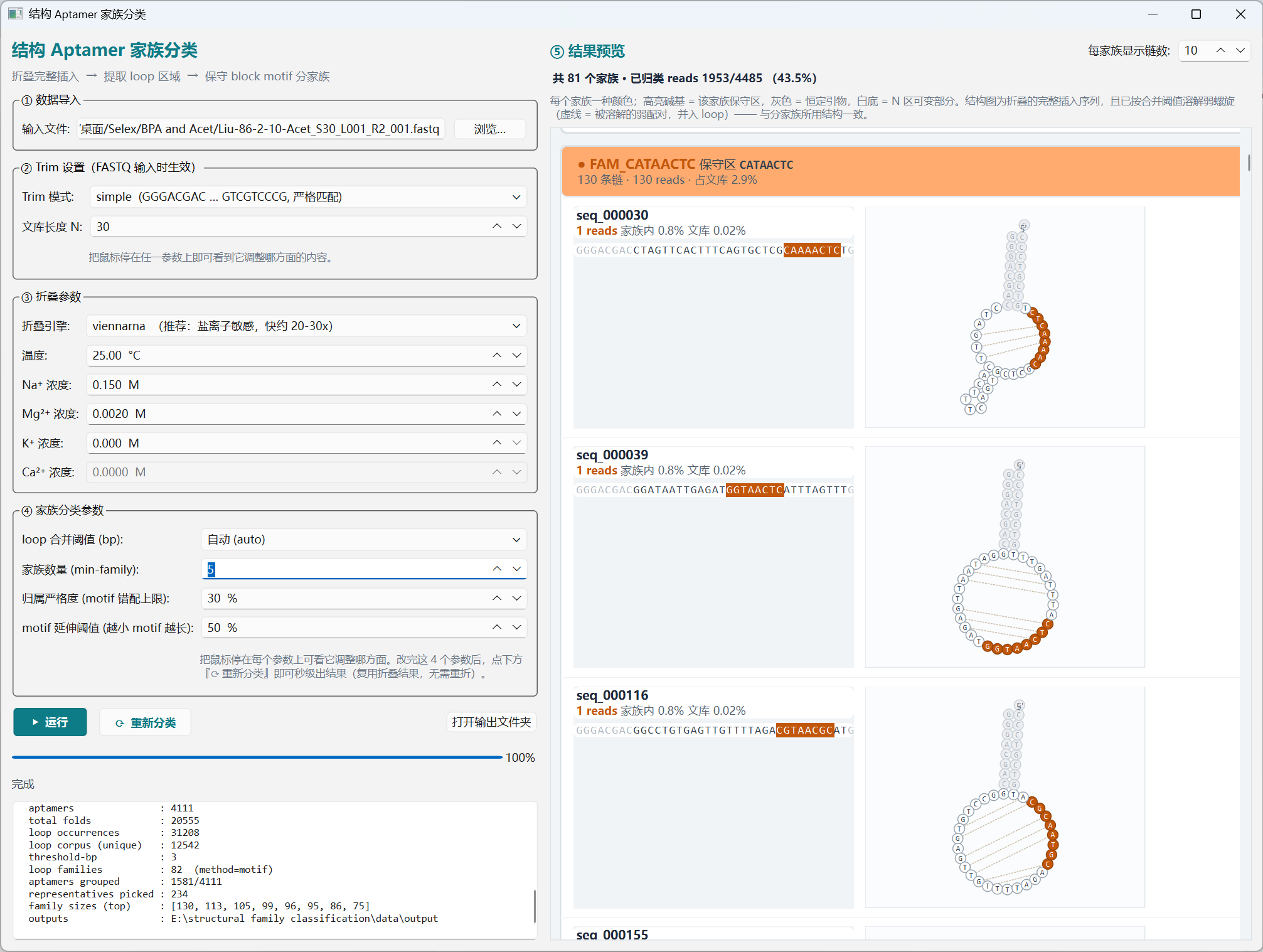
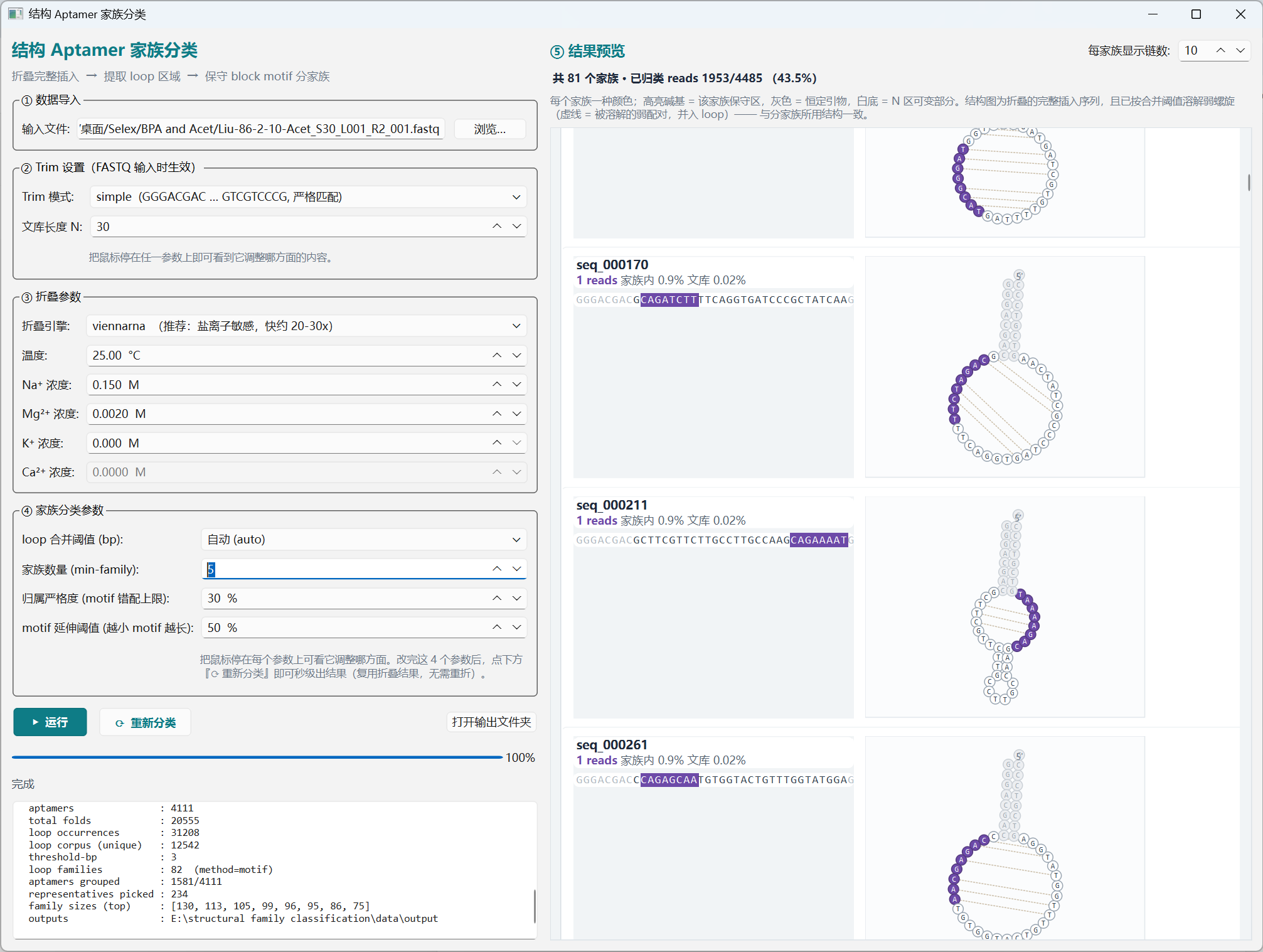
Fig. 04 — Family preview · 家族预览(橙色 motif = FAM_CATAACTC;紫色 motif = 另一家族;每行右侧 = 完整 2D 折叠) -
05
Export representatives for wet-lab
导出家族代表序列做湿实验
07_aptamer_family_bp3.csvassigns every aptamer in the pool to a family.08_family_representatives_bp3.csvpicks one representative per family (ranked by read count) — the shortlist you take into CD / ITC / FRET validation.07_aptamer_family_bp3.csv给文库中每一条适配体分配家族归属。08_family_representatives_bp3.csv按 read 数排序为每个家族选一条代表序列——可直接进入 CD / ITC / FRET 验证的候选清单。
/ Disclaimer · 声明
Things to know.使用前请阅读。
-
Experimental — a preview build, not yet validated.
实验性预览版,尚未完成校准
Loop-centered family assignment is still an engineering hypothesis. Wet-lab validation (CD melting + ITC across representatives of every family from a real SELEX pool) is ongoing. The build above (v0.9.4) is a usable preview so you can try the pipeline on your own pools — but treat its families as a structure-aware shortlist for triage, not a calibrated, final answer.
环路中心的家族归属目前仍是工程假设。基于真实 SELEX 文库每个家族代表的 CD 熔解 + ITC 验证正在进行中。上方的 v0.9.4 是可用的预览版,方便你在自己的文库上试跑流程——但请把它给出的家族视为结构层面的初筛候选,而非已校准的最终结论。
-
Folding inherits AptaFold's limits; the classifier is new.
折叠继承 AptaFold 的限制;分类引擎是全新的
The folding stage reuses AptaFold, so all of AptaFold's accuracy caveats (uncalibrated divalent-cation treatment, experimental engines) apply to the structures this tool classifies. The motif-block classifier, by contrast, is written from scratch for this pipeline — it is not the Aptamer Analysis Tool's k-mer / DBSCAN engine, and its loop-family assignments are themselves the experimental part still under validation.
折叠阶段复用 AptaFold,因此 AptaFold 的全部精度说明(未校准的二价阳离子处理、实验性引擎)对本工具所分类的结构同样适用。而 motif 区块分类引擎是为本流水线从零编写的——它不是 Aptamer Analysis Tool 的 k-mer / DBSCAN,其环路家族归属本身正是仍在验证中的实验部分。
-
Local processing, no telemetry.
本地处理,无遥测
No network calls during the pipeline. Sequences, folds, loops, clusters, and family assignments all live on your machine.
流水线运行期间不发起任何网络请求。序列、折叠、环路、聚类与家族归属全部保留在本机。
-
Will be free for academic use when released.
发布后面向学术免费
Free for academic / non-commercial use, on the same terms as the other Liu Lab tools (MIT-style permissive license). Please point colleagues to this page rather than redistributing the EXE, so they always get the latest preview as the pipeline is calibrated.
学术 / 非商业用途免费,与 Liu Lab 其他工具一致(MIT 风格的宽松许可证)。请把同事指向本页而不是直接转发 EXE,以便他们随流水线校准始终拿到最新预览版。
-
Provided as-is, without warranty.
不提供任何形式的担保
Family assignments are best-effort. Treat them as a structure-aware shortlist for wet-lab triage, not as final answers. Bug reports + early collaboration inquiries: [email protected].
家族归属尽力而为,请将其视为结构层面的湿实验候选清单,而非最终结论。Bug 反馈与早期合作意向请发送至 [email protected]。
/ Acknowledgments · 致谢
Built with the lab.和实验室一起完成。
Developed in the Bionanotechnology & Interfaces Laboratory at the University of Waterloo, led by Prof. Juewen Liu. This pipeline is the natural sequel to AptaFold and the Aptamer Analysis Tool — taking the question "what families are in a SELEX pool?" from sequence-level matching to structure-level matching.
本工具在加拿大滑铁卢大学刘珏文教授的 Bionanotechnology & Interfaces Laboratory 完成。它是 AptaFold 与 Aptamer Analysis Tool 的自然延续——把"SELEX 文库里有哪些家族?"这个问题从序列层面的匹配推进到结构层面的匹配。
-

Prof. Juewen Liu
Advisor · 导师
Professor of Chemistry at the University of Waterloo. Tier 1 Canada Research Chair in Biosensors & Bionanotechnology. Framed the loop-centered hypothesis and provides the SELEX pools and wet-lab validation pipeline that the families are tested against.
-

Xiaohan Zhang
Author · Pipeline & bridge code · 作者 · 流水线开发
Postdoctoral researcher at the University of Waterloo under Prof. Juewen Liu. Designed the loop-extraction + weak-helix-dissolution bridge, the GUI, and the family-assignment join — wiring AptaFold and the Aptamer Analysis classifier into one structure-aware pipeline.
What is reused, and what is new. Only the folding stage is borrowed. Everything downstream — loop extraction, weak-helix dissolution, the motif-block classifier, and the dual-anchor / rescue post-processing — was written from scratch for this pipeline.
哪些是复用、哪些是全新。只有折叠阶段是借用的。其后的全部环节——环路提取、弱螺旋融合、motif 区块分类器,以及双锚点合并 / 救回后处理——都是为本流水线从零编写的。
-
Folding — reused from AptaFold
折叠阶段(复用 AptaFold)
Suboptimal folding via AptaFold's
enumerate_seqfold_suboptimals()— up to 25 structures per sequence within4 kcal/molof the MFE, ranked by ΔG. This is the only borrowed stage; the classifier never recomputes a fold.复用 AptaFold 的
enumerate_seqfold_suboptimals()提供次优折叠——每条序列在 MFE 上下4 kcal/mol内最多 25 个结构,按 ΔG 排序。这是唯一借用的阶段;分类器不重做折叠。 -
Motif-block classifier — written from scratch
motif 区块分类器(完全自写 · 核心创新)
The core engine is original to this project, not the Aptamer Analysis Tool's k-mer / DBSCAN. It mines enriched k-mer cores, grows each into a consensus block, and names a family by the block combination a loop carries — with strict full-consensus membership and no N×N distance matrix. Two original post-processing passes follow: a dual-anchor merge (fuses families that share both a 5′ and a 3′ framework with a variable middle) and an ungrouped rescue (recovers near-identical high-read winners that fall below the motif-seeding threshold).
核心引擎为本项目原创,并非 Aptamer Analysis Tool 的 k-mer / DBSCAN。它挖掘富集的 k-mer 核心、生长为共识区块,以一条环路携带的区块组合命名家族——采用严格的完整共识成员判定,且无 N×N 距离矩阵。其后还有两个原创后处理:双锚点合并(融合共享 5′ 与 3′ 框架、仅中间可变的家族)与未分组救回(救回低于 motif 播种阈值的近乎相同的高 reads 优势序列)。
-
seqfold & ViennaRNA
底层折叠库
The two folding engines underneath AptaFold. ViennaRNA is the default in this pipeline for salt sensitivity and runtime; seqfold is the pure-Python fallback when ViennaRNA is unavailable.
AptaFold 背后两套折叠引擎。本流水线默认使用 ViennaRNA(盐敏感、速度快);当 ViennaRNA 不可用时回退到纯 Python 的 seqfold。
Open-source libraries. The pipeline runs on top of these projects — thanks to their maintainers:
开源依赖。本流水线建立在以下项目之上,感谢各位维护者:
- seqfold
- ViennaRNA
- NumPy
- matplotlib
- PySide6
The motif-block classifier itself uses only the Python standard library — no clustering dependency. · 分类引擎本身仅依赖 Python 标准库,无聚类库依赖。
/ Also in the toolkit · 工具集
Other tools.同系列工具。
Aptamer Analysis Tool
适配体序列分析工具
Sibling SELEX tool — sequence-level family clustering by k-mer + DBSCAN. This pipeline takes the loop-family question its own way, with a self-written motif classifier.
AptaFold
适配体二级结构预测
Four-cation aware DNA aptamer folding. Provides the suboptimal-folding engine this pipeline reuses.
Kd Fitting Tool
Kd 拟合工具
Quadratic binding-model Kd fitting from concentration / signal data. Browser-only, no upload.